

Limites rígidos de impurezas de nitrosaminas em medicamentos são cruciais para proteger a segurança do consumidor. O FDA divulgou importantes orientações e informações sobre o controle de impurezas de nitrosamina em drogas humanas, incluindo principais causas, limites aceitáveis e recomendações para fabricantes de API e fabricantes de medicamentos.
O FDA fez essa determinação devido à importância de fornecer informações importantes aos fabricantes sobre avaliações de risco, testes e outras ações apropriadas que devem tomar para reduzir e mitigar impurezas de nitrosamina em ingredientes farmacêuticos ativos (APIs) e medicamentos. Este documento de orientação está sendo implementado imediatamente, mas continua sujeito a comentários de acordo com as boas práticas de orientação da Agência.
O termo API usado ao longo do texto refere-se a substância medicamentosa, o ingrediente ativo em um medicamento.
O guia publicado pelo FDA recomenda etapas que os fabricantes de APIs e medicamentos devem seguir para detectar e prevenir níveis inaceitáveis de impurezas de nitrosamina em produtos farmacêuticos. O guia também descreve as condições que podem introduzir impurezas de nitrosamina.
A recente descoberta inesperada de impurezas de nitrosamina, que são prováveis carcinógenos humanos, em drogas como bloqueadores do receptor de angiotensina II (ARBs), ranitidina, nizatidina e metformina, tornou clara a necessidade de uma estratégia de avaliação de risco para potenciais nitrosaminas em medicamentos e qualquer produto farmacêutico em risco por sua presença. Este documento revisa a orientação de mesmo título emitida em setembro de 2020 e também estende o período de preparação das avaliações de risco iniciais para 31 de março de 2021.
Descoberta de Nitrosaminas em Medicamentos
A descoberta de nitrosaminas em medicamentos levou o FDA e outros reguladores internacionais a realizar uma análise detalhada dessas impurezas em APIs e medicamentos afetados. Com base no entendimento atual da Agência, esta orientação discute as causas potenciais da formação de nitrosaminas e aconselha os fabricantes de APIs e medicamentos que devem realizar avaliações de risco de seus produtos aprovados ou comercializados e produtos com aplicações pendentes e tomar as medidas adequadas para reduzir ou prevenir o presença de nitrosaminas em medicamentos e IFAs.
Embora impurezas de nitrosamina tenham sido encontradas em apenas alguns medicamentos, e lotes desses produtos tenham sido recolhidos quando havia níveis inaceitáveis dessas impurezas, impurezas de nitrosamina podem existir em outros APIs e medicamentos devido ao uso de processos e materiais vulneráveis que podem produzir impurezas de nitrosamina.
Portanto, as recomendações feitas neste guia se aplicam a todos os APIs sintetizados quimicamente. Eles também se aplicam a medicamentos contendo APIs sintetizados quimicamente e a medicamentos em risco devido a outros fatores descritos neste guia, e não apenas aos medicamentos que foram identificados em anúncios da FDA.
Em geral, os documentos de orientação da FDA não estabelecem responsabilidades legalmente executáveis. Em vez disso, as orientações descrevem o pensamento atual da Agência sobre um tópico e devem ser vistas apenas como recomendações, a menos que requisitos regulamentares ou estatutários específicos sejam citados.
O uso da palavra deveria nas orientações da Agência significa que algo é sugerido ou recomendado, mas não obrigatório.
O FDA tem investigado a presença de impurezas de nitrosamina em certos medicamentos. Desde 2018, vários medicamentos, incluindo ARBs, ranitidina, nizatidina e metformina, foram encontrados para conter níveis inaceitáveis de nitrosaminas.
Em junho de 2018, o FDA foi informado da presença de uma impureza identificada como Nnitrosodimetilamina (NDMA) no ARB valsartan.
Por meio de investigação, a Agência determinou que vários lotes de valsartan e alguns outros medicamentos ARB de diferentes fabricantes continham níveis inaceitáveis de nitrosaminas em medicamentos. Os fabricantes de medicamentos recordaram voluntariamente os lotes afetados desses medicamentos, o que levou a uma escassez de medicamentos em alguns dos produtos afetados. Além disso, a FDA avaliou processos que usam aminas comuns na síntese de API e aprendeu que as vias sintéticas comuns também podem introduzir outros tipos de impurezas de nitrosamina além do NDMA.
Em setembro de 2019, o FDA soube que algumas azia comuns em produtos (ranitidina, comumente conhecida como Zantac, e nizatidina, comumente conhecida como Axid) continham níveis inaceitáveis de NDMA.
A FDA recomendou que os fabricantes retirassem voluntariamente os produtos de ranitidina e nizatidina com níveis de NDMA acima do que a Agência considera aceitável. Recentemente, descobertas preliminares de testes de estabilidade da FDA levantaram preocupações de que os níveis de NDMA em alguns produtos de ranitidina armazenados em temperatura ambiente podem aumentar com o tempo para níveis inaceitáveis.
Os resultados preliminares da FDA usando testes de estabilidade acelerados demonstraram que níveis elevados de NDMA foram medidos em todos os produtos após 2 semanas. Os testes da FDA sugerem que os níveis de NDMA
aumentar com o tempo de armazenamento. Em 1 de abril de 2020, o FDA solicitou que todos os produtos de ranitidina fossem retirados do mercado dos EUA.
Em dezembro de 2019, o FDA tomou conhecimento de que alguns medicamentos de metformina para diabetes em outros países tinham NDMA. À luz dessas informações, a FDA adquiriu amostras de metformina para testar o NDMA. Em fevereiro de 2020, a Agência identificou NDMA em algumas amostras, mas não encontrou níveis que excediam o limite de ingestão aceitável. Em maio de 2020, outros testes da FDA revelaram que certos lotes de formulação de liberação prolongada de metformina continham NDMA acima do limite de ingestão aceitável recomendado pela Agência. Com base nesse teste, a FDA solicitou que os candidatos identificados retirassem voluntariamente esses lotes da metformina de liberação prolongada. A FDA continua a investigar possíveis impurezas de NDMA na metformina e em outros medicamentos e aconselhará as empresas sobre as ações apropriadas.
Como o problema de impureza de nitrosamina se estende além do fornecimento de drogas dos EUA, o FDA e outras autoridades regulatórias fizeram parceria para compartilhar informações, coordenar esforços de inspeção, comunicar métodos analíticos eficazes para detectar e identificar várias nitrosaminas em medicamentos e desenvolver soluções rápidas para garantir a segurança e a qualidade de o suprimento de drogas.
Impurezas de Nitrosamina
O termo nitrosamina descreve uma classe de compostos com a estrutura química de um grupo nitroso ligado a uma amina (R1N (-R2) -N = O), como mostrado na Figura 1. Os compostos podem se formar por uma reação nitrosante entre aminas (secundária , aminas terciárias ou quaternárias) e ácido nitroso (sais de nitrito em condições ácidas).
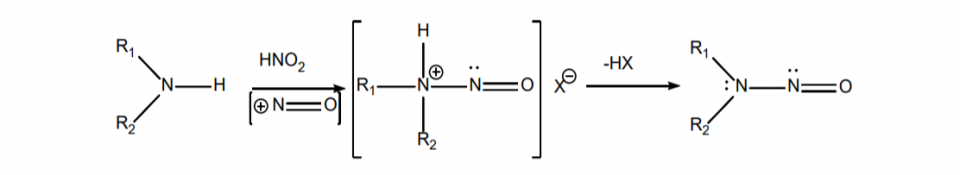
O FDA identificou sete impurezas de nitrosamina que teoricamente poderiam estar presentes em medicamentos: NDMA, N-nitrosodietilamina (NDEA), ácido N-nitroso-N-metil-4-aminobutanóico (NMBA), N-nitrosoisopropiletilamina (NIPEA), N- nitrosodiisopropilamina (NDIPA), Nnitrosodibutilamina (NDBA) e N-nitrosometilfenilamina (NMPA). Cinco deles (NDMA, NDEA, NMBA, NIPEA e NMPA) foram realmente detectados em substâncias ou produtos farmacêuticos.
Estruturas químicas de sete impurezas potenciais de nitrosaminas em APIs e medicamentos
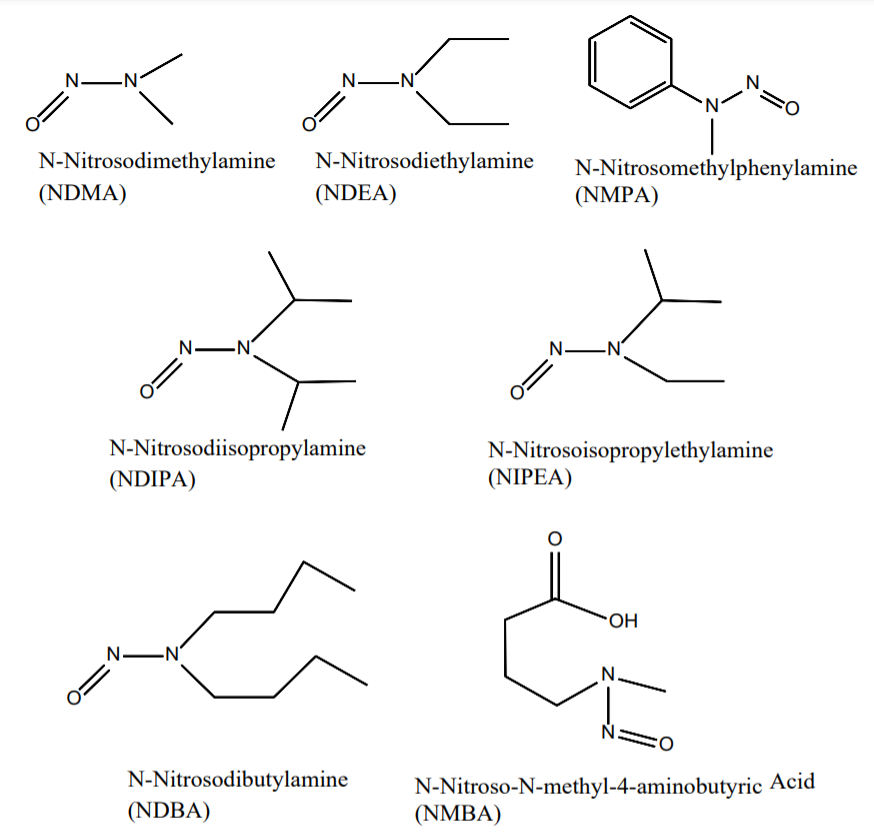
Os compostos de nitrosamina são potentes agentes genotóxicos em várias espécies animais e alguns são classificados como prováveis ou possíveis carcinógenos humanos pela Agência Internacional de Pesquisa sobre o Câncer (IARC). Eles são referidos como compostos de “coorte de preocupação” na orientação do ICH para avaliação e controle M7 (R1) da indústria de impurezas reativas ao DNA (mutagênicas) em produtos farmacêuticos para limitar o risco potencial de câncer (março de 2018).
A orientação recomenda o controle de qualquer carcinógeno mutagênico conhecido, como compostos nitroso, em ou abaixo de um nível tal que haveria um risco insignificante de câncer humano associado à exposição a impurezas potencialmente mutagênicas. Após a descoberta de contaminantes de nitrosamina nos ARBs, o FDA publicou limites aceitáveis provisórios para essas impurezas.
O FDA recomendou que os fabricantes tomem medidas para quantificar os níveis de nitrosamina em seus medicamentos e para reduzir ou remover essas impurezas quando acima do limite provisório; A FDA usou os limites provisórios para orientar a tomada de decisão imediata para avaliação adicional e recalls de produtos, ao mesmo tempo em que equilibra os riscos da potencial exposição a carcinógenos de longo prazo com a interrupção do manejo clínico dos pacientes.
Causas básicas para presença de impurezas de nitrosaminas em APIs
Informações recentes coletadas pelo FDA sugerem várias causas gerais da presença de impurezas de nitrosamina em APIs:
Condições gerais que levam à formação de nitrosamina
A formação de nitrosaminas é possível na presença de aminas secundárias, terciárias ou quaternárias e sais de nitrito em condições de reação ácidas. Nessas condições, os sais de nitrito podem formar ácido nitroso, que pode reagir com uma amina para formar uma nitrosamina.
Há um risco maior de formação de nitrosamina se o ácido nitroso for usado para extinguir a azida residual (um reagente comumente usado na formação do anel de tetrazol ou na introdução do grupo funcional da azida em uma molécula) na presença de aminas precursoras.
Os nitritos usados como reagentes em uma etapa podem ser transportados para as etapas subsequentes, apesar das operações de purificação, e reagir com aminas para gerar impurezas de nitrosamina. Portanto, sempre que os sais de nitrito estiverem presentes, o transporte para as etapas subsequentes não pode ser descartado. Em geral, os processos que usam nitritos na presença de aminas secundárias, terciárias ou quaternárias correm o risco de gerar impurezas nitrosaminas.
Fontes de aminas secundárias, terciárias e quaternárias que podem formar nitrosaminas. As aminas podem estar presentes em um processo de fabricação por uma variedade de razões. O API (ou degradantes API), intermediários ou materiais de partida podem conter grupos funcionais de amina secundária ou terciária. As aminas terciárias e quaternárias também podem ser adicionadas intencionalmente como reagentes ou catalisadores.
Todos esses tipos de aminas podem reagir com o ácido nitroso ou outros agentes nitrosantes para formar nitrosaminas.
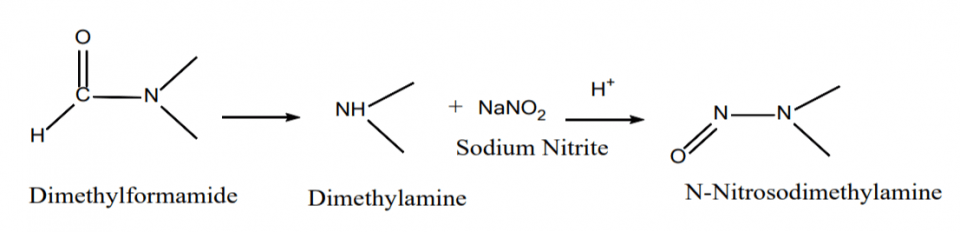
Os solventes de amida, que são suscetíveis à degradação sob certas condições de reação, são outra fonte de aminas secundárias. Por exemplo, sob altas temperaturas de reação por um período de reação prolongado, N, N-dimetilformamida pode se degradar em dimetilamina, que pode reagir com ácido nitroso para formar NDMA. N-metilpirrolidona, N, N-dimetilacetamida e N, N-dietilacetamida também têm vias de degradação semelhantes para formar aminas secundárias que podem reagir com o ácido nitroso para formar impurezas de nitrosamina.
As aminas secundárias também podem estar presentes como impurezas em solventes de amida. Por exemplo, a dimetilamina, que pode reagir com o ácido nitroso para formar NDMA, pode existir como uma impureza em N, N-dimetilformamida.
Formação de NDMA a partir de N, N-Dimetilformamida
As aminas terciárias e quaternárias usadas como reagentes na síntese de APIs podem conter outras impurezas de amina. Demonstrou-se que as aminas terciárias, como a trietilamina, contêm baixos níveis de outras aminas secundárias (como a dipropilamina e a isopropiletilamina). As aminas secundárias e terciárias podem estar presentes como impurezas ou degradantes formados pela desalquilação de aminas quaternárias.
Por exemplo, um catalisador de transferência de fase comum, brometo de tetrabutilamônio, pode conter impurezas de tributilamina e dibutilamina. O nível de impureza de amina que pode levar à contaminação por nitrosamina do API depende do processo e deve ser determinado por cada fabricante de API.
Esta lista das fontes mencionadas não é exaustiva, pois os reagentes de amina podem ser usados para mediar uma ampla gama de transformações sintéticas. Os fabricantes devem avaliar outros reagentes contendo grupos funcionais de amina quanto ao risco potencial de formação de nitrosaminas em medicamentos.
Contaminação em matérias-primas fornecidas pelo fornecedor
As impurezas de nitrosamina podem ser introduzidas quando os materiais do fornecedor, incluindo matérias-primas e matérias-primas, estão contaminados. A Agência observou as seguintes contaminações devido a esta causa raiz:
• A contaminação por nitrosamina ocorreu quando solventes novos (orto-xileno, tolueno e cloreto de metileno) foram contaminados durante o envio dos fornecedores (por exemplo, durante a transferência
entre recipientes de armazenamento).
• O nitrito de sódio é uma impureza conhecida em alguns materiais iniciais (como a azida de sódio) e pode estar presente e reagir com aminas em condições ácidas para formar nitrosaminas. Matérias-primas contendo nitrato, como nitrato de potássio, podem conter impurezas de nitrito.
A quantidade de impureza de nitrito que pode ser tolerada depende do processo e deve ser determinada por cada fabricante de API.
• As aminas secundárias ou terciárias foram relatadas como impurezas em algumas matérias-primas (consulte os detalhes na seção II.B.2 deste guia) e em solventes novos, como o tolueno.
• Os materiais iniciais ou intermediários terceirizados podem estar em risco por contaminação cruzada se forem fabricados em locais onde as impurezas de nitrosamina são produzidas em outros processos.
A conscientização da cadeia de suprimentos de matérias-primas é um fator importante na prevenção da contaminação. Por exemplo, os produtores de API podem não estar cientes da contaminação por nitrosamina em matérias-primas ou matérias-primas que adquiriram de fornecedores; um produtor cujo processo de fabricação não é normalmente suscetível à formação de nitrosamina pode não perceber que o material de origem do fornecedor pode ter tido impurezas introduzidas durante a produção ou transporte.
Solventes, catalisadores e reagentes recuperados como fontes de contaminação
Materiais recuperados como solventes, reagentes e catalisadores podem representar um risco de impurezas de nitrosamina devido à presença de aminas residuais (como trimetilamina ou diisopropiletilamina). Se o processo de recuperação envolver uma etapa de extinção (isto é, ácido nitroso usado para decompor a azida residual), as nitrosaminas podem se formar durante a recuperação do solvente. Essas nitrosaminas podem ser arrastadas se tiverem pontos de ebulição ou propriedades de solubilidade27 semelhantes aos materiais recuperados, dependendo de como ocorre a recuperação e a purificação subsequente (por exemplo, lavagens aquosas ou destilação). Isso aumenta ainda mais o risco de contaminação na recuperação do material. Por essas razões, alguns medicamentos que usam APIs fabricados por certos processos de “baixo” risco foram considerados contaminados. A Agência observou as seguintes contaminações devido a esta causa raiz:
Um local de fabricação pode produzir o mesmo API por mais de um processo sintético que usa solventes comuns. Se algum desses processos sintéticos produzir nitrosaminas ou contiver aminas precursoras, os solventes enviados para recuperação estão em risco. O uso de solventes recuperados provenientes de diferentes processos ou através de linhas de fabricação sem controle e monitoramento pode introduzir impurezas de nitrosamina. Se um solvente recuperado for contaminado dessa forma e, em seguida, usado para fabricar um API, o API será contaminado mesmo se a rota sintética não for normalmente suscetível à formação de nitrosamina.
• A recuperação de matérias-primas (por exemplo, solventes, reagentes e catalisadores) é frequentemente terceirizada para contratados terceirizados. A terceirização de processos pode representar um risco se a instalação de recuperação de terceiros não receber informações específicas suficientes sobre o conteúdo dos materiais que estão processando e se basear apenas em processos de recuperação de rotina.
• As matérias-primas podem ser contaminadas se a limpeza adequada do equipamento entre clientes, ou entre materiais diferentes, não for realizada ou não for validada como capaz de remover cada impureza de interesse. Foi relatado que orto-xileno e tolueno foram contaminados durante a recuperação devido à limpeza inadequada e ao uso de equipamento de armazenamento compartilhado entre diferentes clientes. Procedimentos de limpeza inadequados e não validados também podem levar à contaminação cruzada se as precauções para evitar a contaminação por nitrosamina não forem adotadas antes que os materiais de diferentes clientes sejam combinados para recuperação. Por exemplo, o catalisador cloreto de triN butilestanho (usado como fonte de azida tri-N-butilestanho) foi contaminado em uma instalação terceirizada devido à combinação deste catalisador de diferentes clientes.
Processo como fonte de contaminação por nitrosaminass em medicamentos
Existe o risco de formação de nitrosamina quando uma etapa de dissolução é realizada diretamente na mistura de reação principal (isto é, quando ácido nitroso é adicionado à mistura de reação para decompor a azida residual). Isso permite que o ácido nitroso entre em contato direto com aminas residuais nas matérias-primas usadas no processo de fabricação. As impurezas de nitrosamina podem ser transportadas para as etapas subsequentes se não houver operações adequadas de remoção ou purificação no local, ou se as operações não forem otimizadas para a remoção de impurezas específicas de interesse. Isso pode contaminar todo o processo posterior, uma vez que sejam introduzidos. Mesmo se o processo de têmpera for conduzido fora da mistura de reação principal (consulte a seção II.B.4 neste guia), há um risco de materiais recuperados contaminados serem introduzidos no processo principal.
Falta de otimização e controle de processos
Outra fonte potencial de formação de impurezas de nitrosamina é a falta de otimização do processo de fabricação de APIs quando as condições de reação, como temperatura, pH ou a sequência de adição de reagentes, intermediários ou solventes, são inadequadas ou mal controladas. O FDA observou casos em que as condições de reação variaram amplamente entre os lotes e até mesmo entre diferentes equipamentos de processamento na mesma instalação para o mesmo API.
As múltiplas causas da contaminação por nitrosamina listadas acima podem ocorrer no mesmo processo de API. Portanto, várias estratégias podem ser necessárias para identificar todas as fontes potenciais de contaminação. Os testes de rotina típicos (por exemplo, HPLC) para pureza API, identidade e impurezas conhecidas são improváveis de detectar a presença de impurezas de nitrosamina. Além disso, cada modo de falha pode resultar em diferentes nitrosaminas e em várias quantidades em lotes do mesmo processo e do mesmo produtor de API, com contaminação detectada em alguns lotes, mas não em todos.
Impurezas da nitrosaminas em medicamentos de fontes diferentes da contaminação por API
Os nitritos são impurezas nitrosantes comuns que foram relatadas em muitos excipientes em níveis de ppm. As impurezas de nitrito são encontradas em uma variedade de excipientes comumente usados, que podem levar à formação de impurezas de nitrosamina em medicamentos durante o processo de fabricação do medicamento e o período de armazenamento. O programa de qualificação do fornecedor deve levar em consideração que as impurezas de nitrito variam entre os lotes de excipientes e podem variar de acordo com o fornecedor. Os fabricantes de medicamentos também devem estar cientes de que as impurezas de nitrito e nitrosamina podem estar presentes na água potável.
Alguns medicamentos podem sofrer vias de degradação que formam impurezas de nitrosamina; isso pode ocorrer potencialmente durante o armazenamento do medicamento.
Recomendações sobre Nitrosaminas em Medicamentos segundo a FDA
Como as nitrosaminas em medicamentos são prováveis ou possíveis carcinógenos humanos, a FDA recomenda que os fabricantes considerem as causas potenciais da formação de nitrosaminas descritas neste guia, bem como quaisquer outras vias observadas, e avaliem o risco de contaminação ou formação de nitrosaminas em seus APIs e medicamentos.
Os fabricantes devem priorizar a avaliação de APIs e medicamentos com base em fatores como dose máxima diária, duração do tratamento, indicação terapêutica e número de pacientes tratados.30 Conforme novas informações se tornam disponíveis e o entendimento do FDA sobre nitrosaminas em medicamentos evolui, a Agência pode recomendar que certos medicamentos se tornam prioridades mais altas para avaliação de risco.
Os fabricantes devem consultar a orientação do ICH para gerenciamento de riscos de qualidade da indústria (junho de 2006) para obter detalhes relacionados à identificação, análise e gerenciamento de riscos de qualidade. Os fabricantes de APIs e medicamentos devem tomar medidas apropriadas para prevenir níveis inaceitáveis de impurezas de nitrosamina em seus produtos.
Limites de aceitáveis de impurezas de Nitrosaminas em Medicamentos
A FDA recomenda os seguintes limites de ingestão aceitável (AI) para as impurezas de nitrosamina NDMA, NDEA, NMBA, NMPA, NIPEA e NDIPA. Recomendamos ainda que os fabricantes usem esses AIs ao determinar os limites de impurezas de nitrosamina em APIs e medicamentos.
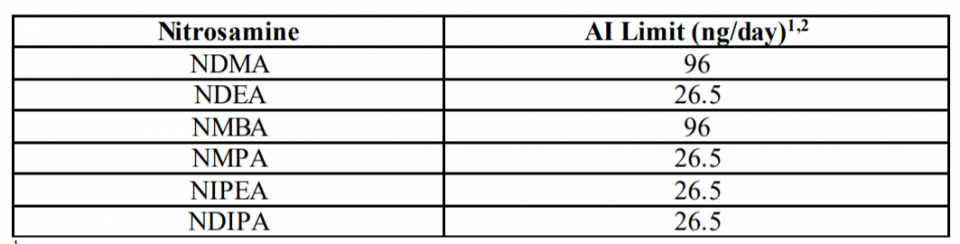
O limite AI é uma exposição diária a um composto como NDMA, NDEA, NMBA, NMPA, NIPEA ou NDIPA que se aproxima de um risco de câncer de 1: 100.000 após 70 anos de exposição. O Apêndice B inclui uma descrição da derivação AI para NDMA, que é um exemplo de como a FDA aplicou ICH M7 (R1) para definir um limite.
A conversão do limite de IA em ppm varia por produto e é calculada com base na dose diária máxima de um medicamento (MDD), conforme refletido no rótulo do medicamento (ppm = AI (ng) / MDD (mg)).
Esses limites são aplicáveis apenas se um medicamento contém uma única nitrosamina. Se mais de uma das impurezas de nitrosamina identificadas na Tabela 1 for detectada e a quantidade total de impurezas de nitrosamina exceder 26,5 ng / dia (o AI para as nitrosaminas mais potentes) com base na dose diária máxima (MDD), o fabricante deve entrar em contato com o Agência de avaliação.
Para medicamentos com um MDD inferior a 880 mg / dia, um limite recomendado para nitrosaminas totais de 0,03 ppm não é superior a 26,5 ng / dia e é considerado aceitável. Para medicamentos com MDD acima de 880 mg / dia, o limite de nitrosaminas totais deve ser ajustado de forma a não ultrapassar o limite recomendado de 26,5 ng / dia.
Se nitrosaminas sem limites de IA publicados forem encontrados em medicamentos, os fabricantes devem usar a abordagem descrita em ICH M7 (R1) para determinar o risco associado à nitrosamina e entrar em contato com a Agência sobre a aceitabilidade de qualquer limite proposto.
Geralmente, métodos sensíveis com limites de quantificação (LOQ) na faixa de partes por bilhão (ppb) são necessários para atender aos baixos AIs recomendados para nitrosaminas. Os fabricantes de APIs e medicamentos devem usar métodos com LOQs iguais ou inferiores a 0,03 ppm.
Os fabricantes devem estabelecer métodos para os quais o LOQ e o limite de detecção (LOD) são tão baixos quanto razoavelmente práticos para produtos para os quais a dose diária máxima é alta (por exemplo, superior a 1 g). Se mais de uma nitrosamina listada na Tabela for detectada, o método analítico deve ser validado para LOQs abaixo de 0,03 ppm para quantificar com precisão um nível de nitrosamina total de não mais do que 26,5 ng / dia. Por exemplo, se o MDD for 1200 mg, o LOQ deve estar abaixo de 0,02 ppm. A página pública da FDA inclui métodos de teste analítico validados recomendados para detectar impurezas de nitrosamina em vários APIs e produtos diferentes.
Os fabricantes de APIs e medicamentos devem seguir as seguintes etapas para mitigar as impurezas de nitrosamina em seus produtos:
- Avalie o risco de impurezas de nitrosamina em APIs, produtos comercializados e produtos sob aplicações aprovadas e pendentes. As avaliações de risco devem ser conduzidas em tempo hábil com base na priorização dos medicamentos. Os fabricantes não precisam enviar documentos de avaliação de risco à Agência, mas devem guardar esses documentos para que estejam disponíveis se solicitados.
- Realizar testes de confirmação39 quando houver risco de presença de impurezas nitrosaminas. Devido às propriedades físico-químicas das nitrosaminas (baixos pesos moleculares, alguma volatilidade e alta toxicidade), os métodos analíticos para nitrosaminas precisam ter especificidade, excelente separação cromatográfica e capacidade de detecção altamente sensível.
- Relate as alterações implementadas para prevenir ou reduzir as impurezas de nitrosamina em APIs e medicamentos ao FDA. Isso inclui o envio de quaisquer alterações do arquivo mestre de drogas (DMF) de acordo com CFR 314.420 (c) e alterações nos aplicativos aprovados conforme exigido em 21 CFR 314,70 e 314,97 e pedidos pendentes em 21 CFR 314,60 e 314,96.
Recomendações para fabricantes de API
Embora não se espere que as nitrosaminas se formem durante a fabricação da grande maioria dos APIs, todos os fabricantes de APIs sintetizados quimicamente devem tomar ações apropriadas para mitigar o risco de contaminação por nitrosaminas para APIs onde há potencial para impurezas de nitrosamina.
Os fabricantes de API devem revisar seus processos de fabricação de API e realizar avaliações de risco para identificar o potencial de impurezas de nitrosamina. Se um risco de impurezas de nitrosamina for identificado, testes de confirmação de lotes devem ser conduzidos usando métodos sensíveis e devidamente validados. Se a avaliação de risco determinar que não há potencial para impurezas de nitrosamina, não há necessidade de tomar outras medidas. Se uma impureza de nitrosamina for detectada, os fabricantes de API devem investigar a causa raiz. Eles devem implementar mudanças no processo de fabricação para reduzir ou prevenir as impurezas da nitrosamina.
Mitigando a presença de impurezas de nitrosamina em APIs
A FDA recomenda que os fabricantes de API realizem as seguintes ações:
Os fabricantes de API devem otimizar o projeto do processo de fabricação de APIs durante o desenvolvimento da rota de síntese (ROS) para minimizar ou prevenir a formação de impurezas de nitrosamina. Os fabricantes de API devem consultar as recomendações em ICH M7 (R1) e as orientações ICH para a indústria Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients (setembro de 2016) e Q11 Development and Manufacture of Drug Substances (novembro de 2012) a este respeito. Os seguintes fatores devem ser considerados durante o desenvolvimento do processo:
- Evitar condições de reação que possam produzir nitrosaminas, sempre que possível; quando não for possível, demonstrar que o processo é adequadamente controlado e é capaz de reduzir consistentemente as impurezas da nitrosamina por meio de estudos de destino e purga apropriados e robustos.
- Usar bases diferentes de aminas secundárias, terciárias ou quaternárias (quando possível) se as condições ROS puderem formar nitrosaminas.
- Ter cuidado quando o ROS envolve o uso de solventes de amida (por exemplo, N, Ndimetilformamida, N, N-dimetilacetamida e N-metilpirrolidona).
- Substituição de nitritos por outros agentes de têmpera para processos de decomposição de azida.
- Otimização e controle consistente das sequências de reações, processos e condições de reação (como pH, temperatura e tempo de reação).
- Projetar um processo de fabricação que facilite a purga de impurezas de nitrosamina nas etapas de processamento subsequentes.
Controle de impurezas de nitrosamina em APIs
Os fabricantes de medicamentos devem realizar avaliações de risco para determinar o potencial de impurezas de nitrosamina em medicamentos. Uma avaliação de risco deve envolver a colaboração com o fabricante do API para auxiliar na identificação do API ROS ou outras condições do processo de fabricação do API que colocam o medicamento em risco de impurezas de nitrosamina. A avaliação de risco também deve incluir a avaliação de qualquer via (incluindo degradação) que pode introduzir nitrosaminas durante a fabricação ou armazenamento do medicamento. Se a avaliação de risco determinar que não há potencial para impurezas de nitrosamina, não há necessidade de tomar outras medidas.
Se um risco de nitrosaminas em um medicamento for identificado, o teste de confirmação dos lotes deve ser conduzido usando métodos sensíveis e devidamente validados. Se uma impureza de nitrosamina for detectada, os fabricantes devem investigar a causa raiz e implementar mudanças no processo de fabricação para mitigar ou reduzir as impurezas de nitrosamina.
Controle de impurezas de nitrosamina em medicamentos
Os fabricantes de medicamentos devem testar amostras representativas de todos os componentes recebidos, incluindo lotes de API em risco, antes do uso, conforme exigido em 21 CFR 211.84.48 Para atender aos regulamentos CGMP em 21 CFR 211 subparte E e ser consistente com a orientação ICH para o Sistema de Qualidade Farmacêutica Q10 da indústria (abril de 2009), os fabricantes de medicamentos devem continuar testando lotes de API até que tenham verificado que o fornecedor de API pode fabricar API de forma consistente sem níveis inaceitáveis de nitrosamina.
Os fabricantes de medicamentos, ao projetar sua estratégia de controle, devem avaliar se os nitritos podem estar presentes durante os processos de fabricação onde APIs de risco são usados. Eles também devem avaliar se as nitrosaminas podem se formar em um medicamento acabado durante a vida útil do medicamento. Se uma nitrosamina for introduzida no medicamento por meio de fontes exógenas que podem ser evitadas, os fabricantes devem eliminar a fonte de contaminação.
Se uma impureza de nitrosamina for detectada acima do LOQ, o fabricante deve desenvolver uma estratégia para garantir que o nível de nitrosamina permaneça dentro do limite de IA. A estratégia de controle deve incluir limites de especificação para as nitrosaminas identificadas. Essa estratégia de controle também é recomendada quando a introdução de nitrosamina é inerente à estrutura do API, ao API ROS ou ao processo de fabricação do API ou medicamento. Dadas as incertezas existentes em relação às impurezas de nitrosamina e sua presença nos medicamentos, o teste de cada lote na liberação deve ser realizado. Abordagens alternativas devem ser apoiadas por suficiente compreensão do processo e evidências de controle estatístico adequado e devem ser submetidas ao FDA em um suplemento antes da implementação.
Se lotes de medicamentos com níveis inaceitáveis de impurezas de nitrosamina já estiverem sendo distribuídos, os fabricantes de medicamentos devem entrar em contato com a FDA para que a Agência possa determinar a ação regulatória para os medicamentos específicos. Qualquer lote de medicamento que contenha níveis de impurezas de nitrosamina iguais ou acima do IA recomendado não deve ser liberado pelo fabricante do medicamento para distribuição. Os fabricantes devem entrar em contato com a Agência se um recall for iniciado. De acordo com a seção 501 da Lei de Alimentos, Medicamentos e Cosméticos (FD&C Act), um medicamento que não seja fabricado, processado, embalado ou mantido em conformidade com CGMP para garantir que atenda a determinados padrões de qualidade e pureza é considerado adulterado. A FDA pode exercer critérios regulatórios quando garantido para prevenir ou mitigar a escassez de um medicamento.
Confira o material original publicado pelo FDA:
Guia de Orientação para controle de impurezas de nitrosamina em medicamentos
Fonte: FDA